
The c-MET/HGF signaling regulates multiple cellular processes that stimulate cell proliferation, invasion and angiogenesis. The c-MET pathway has significant interaction with other signaling pathways. The deregulation of c-MET plays an important role in the formation, growth, maintenance, and invasion of tumors. It is involved in various cancers, including lung, colon, liver and stomach cancer. Many studies on the abnormal signaling of c-MET offer strong reasons to investigate the therapeutic potential of some receptor antagonists. Therefore, more active substances must be selected and integrated effectively into well-designed clinical trials, these include predictive biomarkers such as genetic mutation, amplification and overexpression of immunohistochemical c-MET. The complex interconnection of cell control networks provides new pathways for combining EGFR inhibitors and other targeted drugs. Thus, c-MET can be used as potential biomarkers for cancer therapeutics.
c-MET is a tyrosine kinase receptor in the MET family. Normally, it is expressed on the surfaces of several epithelial cells. HGF/SF is a common ligand for c-MET receptors. Cooper et al. were the first to identify the MET oncogene in 1984. They chemically transformed the human osteocarma cell line by transfection analysis in NIH/3T3 cells and mapped the MET oncogene. The MET oncogene was identified by the 7q21-31 chromosome band, showing a genetic shift between two different loci, MET’s proto-oncogenes are on chromosome 7 and on chromosome 1’s transfer promoter regions (TPRs). After analyzing the translation product of the proto-oncogene 21-exon MET proton, it was discovered on the surface of the cells and classified as a tyrosine kinase growth factor receptor. The MET-proton-oncogene product acts as a receptor for the HGF tyrosine kinase transmembrane. The receptor is mainly present in melanocytes, epithelial cells and epithelial tissues, including the liver, the intestine, the kidneys and other organs. HGF regulates various transmission cascades on the c-MET receptor and acts on c-MET receptors in a paracrine manner (such as mitogenactivated protein kinase), phosphatidylinositol-3 kinase (PI3K)/AKT, and Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways. The c-MET network consists of multiple signal molecules that interact with specific physiological processes. In addition to the properties of microbial, molecular, and morphological, c-MET protects against apoptosis by interactions with PI3K/AKT (phosphate di-tyrosine triphosphate/protein kinase-B). However, the co-expression of c-MET and HGF has been observed in various wound closure, tissue regeneration and embryonic events.
c-MET is a receptor tyrosine kinase that is found primarily in melanocytes, endothelial cells and epithelial tissues such as liver, gastrointestinal tract, and kidneys. In normal cells, primary MET transcripts produce 150kDa peptides and partially glycosylated 170kDa precursor proteins. Originally isolated from the line of human gastric tumor cells, the receptor contained a 50kDa chain and 145kDa chain and was linked to a sulfur bridge in a transmembrane αβ complex of 190 kDa. 170kDa precursor proteins are transformed by glycosylation and subsequent translation to produce mature heterodimers. The α -subunit is located on the outer layer, and the β -string, a single-pass chain, forms the core of the three main regions of the c-MET. The extracellular area of c-MET is highly adapted to HGF while a single transmembrane segment and a cytoplasmic tyrosine kinase domain are cut by juxtamembrane and carboxyend sequences. In the extracellular part, there are three types of domains. The SEMA domain, including 500 amino acids, is based on the α and β subunits in their entirety. The preserved c-MET receptor sequences are structurally similar to large families of ligand receptor pairs with semaphorin and plexin. Semaphorin is a large protein family that helps to decompose cells and induce reprehensible orientation events, such as cell dispersion during neural development. Under the SEMA domain, the PSI domain is a common domain for Plexins, Semaphorins, and Integrins. It is connected to the transmembrane helix through four IPT domains (immunoglobulin-like, plexin-like, transcription factor). HGF can be connected to two locations in the SEMA area, one with a low specificity contact and a high specificity contact, and the other with a low specificity contact and a high affinity contact in the IPT region. The intracellular domain consists of three parts: the Juxtamembrane sequence, which down regulates kinase activity followed by Ser975 phosphorylation in the catalytic region for the activity of kinase leading to transphosphorylation of Tyr1234 and Tyr1235. A dimerization of the receptors is caused by the phosphorylation of the tyrosine residues in the kinase domain. This may lead to the autophosphorylation of bidentate docking sites in the carbonate terminal tail of the cytoplasm (the Tyr1349 and Tyr1356 tyrosine residues). The total length of MET as a proto-oncogene is 125,982 bp and belongs to the 7q31 chromosome. HGF is a family of plasminogen-related growth factors known as PRGF-1. The length of HGF gene spans 70 kb on chromosome 7q21.1. HGF initially in the form of pro-HGF is further cleaved by a protease to mature HGF.
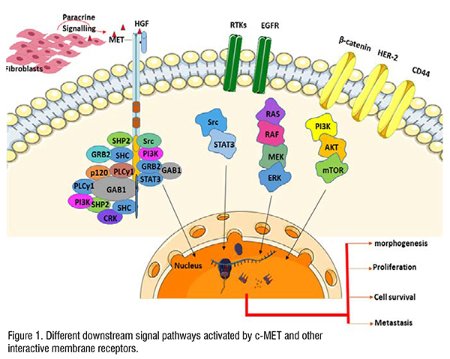
HGF is the main ligand of the c-MET receptor and acts as a biological signal. HGF is produced as an inert precursor of a single chain. The elimination by extracellular protease transforms the predecessor into two-chain functional heterodimers. HGF is mainly distributed in most tissues’ extracellular matrix in inactive forms and is separated from mature forms of proteoglycans such as heparin. HGF originates mainly from mesenchymal cells, which act paracrine in epithelial cells that express c-MET receptors. Several cytokines like interleukin 1, 6, tumor necrosis factor- and the transformation factor- induce transcriptional upregulation, both in HGFs and macrophages, and in c-METs and epithelial cells. Tumor stroma increases the expression of pro-HGF-activated proteins such as plasminogen activation systems and matriptases. Therefore, HGF activates and produces more biological functions by controlling combinations of transcription regulation and post-transcription regulation, which also eventually leads to optimal MET activation on target cells. It is also an important part of the physiological defense mechanism against tissue damage. When HGF is bound, c-MET is activated by the activity of kinase and then the receptor dimerization is completed and followed by transphosphorylation of two catalytic tyrosine residues Tyr1234 and Tyr1235 within the kinase activation region. Subsequently, two additional phosphorylated tyrosine residues are docked in the terminal tail of the carboxylic acid (Tyr1349 and Tyr1356), which, upon activation, can serve as degeneration elements to recruit several other subsequent signaling molecules. MET serves as a substrate for the phosphatase of protein tyrosine, including PTP receptor density-increasing phosphatase 1 (DEP 1) and leukocyte common antigen related (LAR) and the non-receptor PTPs, PTP1B and T cell PTP. These phosphatases may disrupt the c-MET signal by promoting the dephosphorylation of catalytic tyrosine or docking tyrosine. Many scaffold proteins, associated with c-MET receptors, activate downstream signal transmission pathway, this includes cascades of kinase of mitogen-activated proteins (MAPKs), kinases of extracellular signal control 1 (ERK1) and ERK2, kinases of end-amino-terminal Jun (JNKs), and p38, the phosphoinositide 3-kinase–Akt (PI3K–AKT) axis, signal transducer and activator of transcription proteins (STATs), and the nuclear factor-κb inhibitor-α (iκbα)–nuclear factor-κb (NF-κb) complex (Figure1). The C terminal tail of c-MET is linked to a wide range of Src homology domains, including PI3-K, Src non-receptor kinase, and effects the adapters of SHP2 (PTPN11) that connect protein 2 of growth factors receptors (GRB2) and SH2 domains contain converting proteins (SHC).; an upstream activator of Src and ras), phospholipase Cγ1 (PlCγ1) and the transcription factor STAT3. c-MET receptors also provide additional binding sites for other adapter proteins and are associated with the phosphorylation of binding protein 1 (GAB1) associated with GRB2. GAB1 is connected to c-MET receptors through 13 MET binding sites of amino acids and indirectly through GRB2 linked to MET. Some transducer proteins are involved in c-MET receptor signal cascades, mainly in interactions with the GAB1 scaffold adaptor. CD44 is recognized as another protein interacting with c-MET in normal epithelial cells.
c-MET was first discovered in the early 1980s as a product of chromosome rearrangement after a carcinogenic treatment with N-methyl-N′-nitro-N-nitrosoguanidine. This reorganization leads to the constitutive fusion of TPR-MET oncogene, after dimerization, a leucine-zipper motif is formed in the TPR molecule, which is converted to an oncoprotein. This satisfies the structural requirements for the constitutive activity of the c-MET kinase. TPR-MET is capable of transforming epithelial cells and inducing spontaneous mammary tumors when transgenic mice are generally overexpressed. These results set the basis for the ongoing efforts to discover all the cancer-related capabilities of c-MET. More than 10 years of proof of concept of c-MET’s role in human cancer have been required. The discovery of the activation point mutation in the germ line of patients with hereditary renal cell carcinoma was obvious. However, spontaneous MET mutations occur only rarely between 2 and 3 percent. The expression of aberrant MET is widespread in various malignancies, particularly non-small cell lung cancer (NSCLC), gastrointestinal (GI) cancer, and hepatocellular carcinoma (HCC). The interaction between MET and the HER2 family is becoming an important mechanism for the progression of the tumor and treatment resistance. MET signals also interact with vascular endothelial growth factors (VEGFs) and VEGF receptors (VEGFRs). MET activation increases the expression of VEGF-A, and promotes angiogenesis and the growth of endothelial cells.
Many reports show that altered RTK activation levels play an important role in cancer pathophysiology. Deregulation and subsequent aberrant signalling of c-MET can be caused by different mechanisms, including gene amplification, overexpression, activation of mutations, and increased stimulation by autocrine or paracrine ligands, and interaction with other active cell-surface receptors. In preclinical studies, several c-MET small molecule inhibitors and monoclonal antibodies have been evaluated. Studies have shown that the expression of c-MET is related to the expression of immune regulation molecules, such as programmed cell death ligand (PDL-1) and indoleamine-2,3-dioxygenase (IDO) in cancer cells. This assures that c-MET can be effectively targeted for immunotherapy in clinical research. c-MET is also associated with chemotherapy by avoiding traditional clinically suppressive signals such as EGF. Gefitinib and c-MET inhibit additive synergy demonstrated in cell line models. Many studies reported that c-MET was overexpressed in various cancers, including lung, breast, ovaries, kidneys, liver, thyroid and gastric cancer. This excessive expression may be due to transcription activation, hypoxia-induced excessive expression, or MET amplification, especially in a few cancers. It is reported that a transgenic mouse overexpressing c-MET spontaneously develops liver cell carcinoma, and when the transgene was inactivated, tumor regression was reported even in large tumors.
References

Dr. Dhruv Kumar, Senior Associate Professor at the School of Health Sciences and Technology, UPES University, Dehradun, Uttarakhand, India. His lab is currently focusing on Translational Cancer Research, mainly to understand the role of tumor microenvironment in metabolic alterations in solid tumors including Head and Neck, Breast, Brain, Prostate, Pancreatic, Gallbladder and Skin cancer and to understand the genomic and mutational heterogeneity in solid tumors.

Sibi Raj, ICMR-SRF Fellow, currently pursuing PhD in Cancer Biology under the guidance of Dr Dhruv Kumar at the School of Health Sciences and Technology, UPES University, Dehradun. Her research interest is to explore the role of c-MET in metabolic dysregulations in Head and Neck Cancer.






