Operating in one of the most demanding and highly regulated environments, pharmaceutical companies face the constant challenge of managing medicinal product information for regulatory labelling submissions that comply with regional and national agency requirements.
High standards demanded by regulatory agencies during the submissions process – which varies by country – puts enormous pressure on firms to ensure that the filings are effectively coordinated between the central team and local affiliates and that every element of their application is flawless.
The approval process is a long and arduous journey and keeping product information up-to-date in multiple languages requires mobilising many resources centrally and locally. Local regulatory affairs stakeholders may outsource or be directly involved in the translation, review and formatting process. This constant handling of the documentation can cause inconsistencies and quality issues and, most importantly, limit the time to be allocated to main regulatory activities.
As regional and national authorities'requirements evolve and their quality expectations increase, many companies are faced with difficult choices when it comes to developing a multilingual regulatory labelling infrastructure that is scalable, cost efficient and future-proof.
To maximise global reach and reduce time to market, pharmaceutical companies should consider employing new technologies to enhance compliance of their product information with regional and national regulatory filing requirements in multiple languages and geographies.
Label management is complex, spanning the entire lifecycle of a medicinal product, and has global and regional implications. Those involved in label management – from creation through to implementation – work with crossfunctional disciplines within the organisation, e.g. pharmacovigilance, regulatory affairs, medical affairs, and supply chain. It is also one of the most common areas of critical inspection findings by the governing regulatory authorities.

Typical inspection failures include failure to identify and submit safety variations, delays in submitting variations, and failure to implement updated reference safety information following variation approval. Taking into consideration the variation in country-specific regulations, translation requirements and local functions compound these challenges. Pharmaceutical companies have invested heavily in personnel and disparate technologies to manage the end-to-end (E2E) labelling process; however, facing increasing complexity, inherent legacy systems and processes, pharmaceuticals are faced with a significant obstacle to improve this overall process.
The following barriers to making E2E labelling a reality are the most commonly cited:
- Organisational change management: Most stakeholders span the entire organisation and have grown accustomed to working in Microsoft Word and managing information at the file level.
- Siloed technologies: The extremely wide array of systems (RIM, ERP, tracking, etc.) involved across the labelling process, the lack of integration between them and the many manual steps push the boundaries of any centralisation exercise.
- No single source-of-truth: Ensuring that the entire organisation, regardless of geographic location, use current approved content in their local submission remains a challenge.
- Organising complex global regulations: Every country follows very specific guidelines when it comes to labelling format and content.
- Submission delays: Managing new drug registrations, variations, extensions to deadlines, and compliance requirements across multiple markets is a significant challenge. Launching or updating product information simultaneously in multiple countries poses yet another challenge.
- Legacy products and filings: Most organisations retain a wide portfolio of legacy products that were placed in local markets years ago with minimal visibility into these off-patent filings.
- Fragmented collaboration: Ineffective content review and collaboration can jeopardise registrations and submissions. This is particularly crucial for organisations with teams located globally.
With the number of co-development, - licensing and – commercialisation agreements on the rise, most biopharma companies are looking to significantly increase their ability to share labelling content globally. Furthermore, as these organisations are embracing new cloud-based solutions as their authoritative source of content across their entire ecosystem, a structured and global approach to content, or even to data, becomes more of a reality.
Companies follow vastly different paths when it comes to their digitalisation journey and contending with the reality of their global regulatory labelling infrastructure, a controlled and focused step-by-step approach will help establish the necessary foundations for a future E2E labelling model.
Modular and Structured “Intelligent” Content
By leveraging a structured approach to the creation of new labelling content, biopharma companies will be in a position to access a single repository for labelling documents while minimising rework and duplicate information entry. Additionally, with component content management systems (CCMS) providing a “Word-like” authoring environment and focusing on new labelling content first, the negative impact associated with change management and legacy labelling content conversion is minimal. This highly targeted approach would also help further support and validate a compelling ROI, especially when linked to new standards set forth by the regulators, such as the identification of medicinal products (IDMP) ISO standards.
Cloud Assimilation
Interconnectivity between systems (CCMS, TMS, RIM, ERP, etc.) and other data repositories remains the single most important element in any E2E labelling initiative. As such, ensuring that all these technologies are designed with ease of integration in mind will determine the long-term viability of your model. Furthermore, this will prove even more crucial as the biopharma industry further transitions from a content-based to a data-based regulatory content management model. (Figure 1)
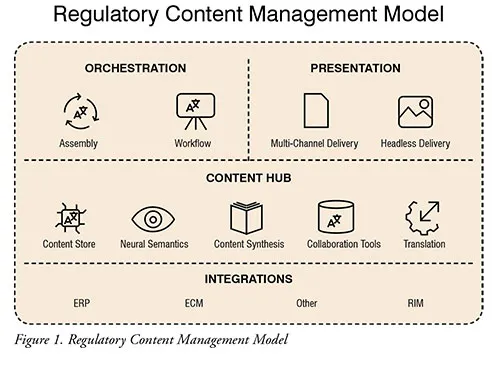
Translation Management System (TMS)
Helping biopharma companies evolve their current translation process to one where translation activities are streamlined and automated represents the single largest opportunity for the industry with the lowest overall risk. By centralising translation requirements into a single cloud-based system, companies can decrease time-to-market and costs, while preserving data integrity and increasing visibility and traceability of their activities on a global scale.
Technology will play an essential role in any E2E labelling initiative but it is not a silver bullet. If there is lack of process standardisation then implementing technology is not going to achieve the desired results. While ensuring that your various systems are interconnected is crucial, one should look at taking advantage of the deployment of these systems to further drive process harmonisation and ease the collaboration between the central team and local affiliates. An organisation can reduce its risk by selecting a part of the process that has defined controls and limited exposure. Risk could be minimised further by developing an approach to efficiently manage translations for a select number of countries or in a given geographic region. Once the model is viable and generates a clear ROI, it will form the basis to further support global expansion.






