
Ensuring quality and regulatory compliance in the biopharmaceutical industry is critical to safeguarding product safety and efficacy. With the increasing complexity introduced by biologic products, which often involve intricate manufacturing processes and sensitive living organisms, the industry faces persistent and evolving challenges in meeting good manufacturing practice (GMP) standards and complying with regulations set by authorities such as the EMA, FDA, and ICH. These challenges are further compounded by the need for continuous innovation and the pressure to bring new therapies to market quickly. Extensive experience in GMP environments, particularly in the QC field in laboratory activities, demonstrates that implementing advanced quality management systems is essential to navigate these complexities, meet stringent regulatory requirements, and provide safe and effective products that patients can rely on (EMA, 2018; ICH, 2020). The cost of non-compliance, both in terms of patient safety and financial impact, underscores the importance of robust quality systems.
What are GMPs? Good manufacturing practices (GMPs) refer to the minimum standards that pharmaceutical manufacturers must adhere to in their manufacturing processes. These practices ensure product quality, safety, and efficacy and are applied by agencies such as the EMA and FDA.
Due to the rapid evolution of biologics, there is a growing need for adaptive quality strategies that respond to emerging manufacturing challenges. Companies must maintain compliance, and in addition implement continuous improvement mechanisms to stay in line with regulatory expectations and technological advances.
Quality Assurance Practices
In biopharmaceutical manufacturing, the integration of comprehensive quality management systems (QMS) and the meticulous implementation of risk-based manufacturing approaches are not merely advisable but critical to achieving and maintaining regulatory compliance. Professionals with significant GMP experience recognize that these systems facilitate an organized and systematic method for the early detection and proactive correction of variations and deviations in both manufacturing and analytical processes, thereby ensuring consistent product quality and minimizing the risk of costly errors or non-compliance issues. An important and indispensable aspect of this practice is the rigorous validation of analytical methods to confirm their accuracy and reliability, alongside strict adherence to GMP standards, which provides a framework for quality at every stage of production. Furthermore, the emphasis on robust documentation practices ensures that all processes are transparent, traceable, and auditable.
Key Elements of a QMS
• Development and control of SOPs
• Implementation of CAPAs
• Internal audits
• Change control processes
• Risk management and training programs
Implementing a risk-based quality assurance system that strategically identifies critical control points throughout the manufacturing process significantly improves compliance outcomes and minimizes the occurrence of noncompliance, thereby safeguarding product integrity and patient safety. Continuous monitoring of key production variables through the utilization of real-time process analytical technology (PAT) systems strengthens the control and optimization of process parameters, enabling timely interventions and adjustments (FDA, 2019).
Compliance Challenges
Maintaining consistent quality and rigorously meeting all regulatory requirements poses significant and multifaceted challenges for biopharmaceutical manufacturers. One of the foremost issues is ensuring data integrity, which is paramount, especially in the context of analytical data management and the complex processes of instrument validation. Every measurement obtained during laboratory equipment validation must be not only accurate but also fully traceable, auditable, and compliant with the stringent requirements of GMP standards (EMA, 2018). This necessitates meticulous record- keeping and robust electronic systems that prevent data manipulation and ensure the reliability of results. The effective management of deviations and out-of-specification (OOS) results is also of critical importance; a well-designed and diligently implemented deviation management system is essential to minimize the potential for human error, facilitate thorough investigations, and ensure rapid and effective problem resolution. Rigorous and systematic investigative processes, coupled with comprehensive and detailed documentation, are indispensable for accurately identifying the root causes of deviations, implementing effective corrective and preventive actions (CAPA), and preventing recurrence.
The growing emphasis on data security, driven by the increasing volume of sensitive information and the need to protect against cyber threats, and the systematic management of regulatory documents, which often involves extensive and complex documentation, necessitates the digitization of information processes and the adoption of robust automated systems to maintain transparency, enhance audit readiness, and streamline regulatory submissions (FDA, 2019).
Common Compliance Risks
• Incomplete documentation
• Lack of audit trail
• Uncontrolled deviations
• Manual data entry errors
Additionally, regulators are increasingly requiring organizations to demonstrate a strong and demonstrable quality culture. This expectation includes transparent and well-documented decision- making processes, the consistent application of thorough root cause analysis methodologies, and a proactive emphasis on preventative actions, all of which must be meticulously documented and readily available for detailed review during regulatory inspections.
Advanced Quality Control Technologies
The strategic application of digital technologies has significantly transformed and continues to revolutionize quality and compliance management within the biopharma sector, offering unprecedented opportunities for enhanced efficiency, accuracy, and control. Artificial intelligence (AI)-driven quality monitoring systems, for example, employ sophisticated machine learning algorithms to continuously analyze vast quantities of production data in real time, enabling the early detection of subtle deviations from expected parameters and the accurate forecasting of potential process variations through the intelligent identification of complex patterns in historical data. This proactive and predictive approach empowers manufacturers to implement timely adjustments, optimize the manufacturing process, reduce variability, and minimize the risk of quality defects (ICH, 2020).
Benefits of EBR Systems
• Real-time access to batch data
• Improved traceability and audit readiness
• Reduction in human error
• Compliance with data integrity requirements
Automated data recording systems, which interface directly and seamlessly with a wide array of laboratory instruments, ensure the efficient and error-free execution of validation procedures, while simultaneously minimizing the potential for manual data entry errors. These systems meticulously record every measurement with complete traceability, creating an immutable audit trail and supporting a robust risk-based quality assurance framework that systematically identifies, assesses, and effectively mitigates potential production hazards (FDA, 2019). The implementation of electronic batch records (EBR) has fundamentally redefined documentation practices in GMP environments, moving away from paper-based systems and towards digital solutions.
EBR systems provide an exact, comprehensive, and readily accessible digital record of each and every step in the manufacturing process, ensuring that all critical process parameters are accurately,
consistently monitored, and readily available for audit purposes, thereby significantly enhancing transparency and facilitating regulatory inspections (EMA, 2018).
Other innovative technologies, such as digital logbooks, cloud-based document management systems, and integrated quality dashboards, further contribute to streamlined compliance monitoring, facilitate real-time decision-making based on accurate and up-to-date information, and improve overall operational efficiency. As technology continues its rapid evolution, the scalability, adaptability, and interoperability of these digital tools will be absolutely central to achieving sustained regulatory success and maintaining a competitive edge in the global biopharmaceutical market.
Strengthening Compliance through International Regulatory Alignment
To ensure consistency and reliability in biopharmaceutical manufacturing, it is essential to align internal practices with international regulatory frameworks. Adherence to globally recognized standards, such as the European Union GMP guidelines (EudraLex Volume 4), the FDA's 21 CFR Part 210 and 211, and the ICH Q10 on Pharmaceutical Quality Systems, supports the development of robust quality systems and facilitates access to international markets.
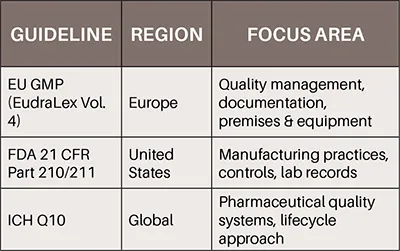
These frameworks not only outline expectations for documentation, validation, and deviation management, but also emphasize the importance of quality lifecycle approaches and continuous process improvement (ICH, 2008; EMA, 2022; FDA, 2023). Integrating such regulatory alignment into daily operations promotes a culture of quality and prepares organizations for successful inspections and audits in all jurisdictions.
Furthermore, cross-functional collaboration is key to embedding international compliance practices. QA, QC, regulatory affairs, production, and IT must work in synergy to interpret, implement, and monitor adherence to these guidelines across departments.
Establishing a Foundation for Quality
The establishment of a robust foundation for quality within a biopharmaceutical organization is paramount to the successful implementation and ongoing effectiveness of its Quality Management System (QMS). This foundational stage necessitates a holistic approach that encompasses several critical elements, beginning with unwavering leadership commitment. This commitment transcends mere resource allocation; it demands that leaders actively champion a quality-focused culture, embedding quality principles into the very fabric of the organization's values and decision-making processes. Leaders must clearly articulate a quality vision, communicate its importance to all personnel, and foster an environment where quality is prioritized over expediency.
Complementing this leadership-driven culture is the development of a comprehensive Quality Plan. This plan serves as a strategic roadmap, outlining the organization's quality objectives, defining key performance indicators (KPIs) to measure progress, and establishing the processes and procedures necessary to achieve these objectives. The Quality Plan must be meticulously documented, regularly reviewed, and adapted to evolving regulatory requirements and business needs.
Finally, adequate resource management is indispensable. This entails not only providing sufficient financial resources but also ensuring the availability of qualified personnel, appropriate equipment, and suitable facilities. Crucially, a strong emphasis must be placed on comprehensive training programs and rigorous competency assessments to ensure that all personnel possess the knowledge, skills, and abilities necessary to perform their assigned tasks effectively and in compliance with GMP standards. This includes initial training, ongoing training, and specialized training for specific roles and responsibilities.
Conclusion
Maintaining robust quality and ensuring unwavering compliance in the biopharma industry requires not only strict adherence to comprehensive regulatory frameworks but also the proactive and continuous adoption of innovative technologies and the implementation of sound and effective management practices. Extensive GMP experience consistently confirms that the strategic deployment of advanced technologies, encompassing AI-driven monitoring systems, automated data management solutions, and real-time process monitoring capabilities, is absolutely critical to significantly enhancing regulatory compliance outcomes, ensuring the integrity and safety of biopharmaceutical products, and ultimately protecting patient well-being. Continued investment in quality culture, personnel development, and cross-departmental integration will be necessary to sustain these improvements over the long term.
As the biopharmaceutical industry continues its rapid evolution, driven by scientific advancements and the development of increasingly complex therapies, the seamless and effective integration of these digital solutions, in close combination with proactive quality management strategies and risk- based methodologies, will play an absolutely pivotal role in successfully addressing the increasingly complex challenges associated with quality assurance, quality control, and regulatory compliance. The ongoing and sustained incorporation of these technological innovations empowers biopharmaceutical professionals to navigate the intricate and ever-changing regulatory landscape more effectively, streamline operations, and contributes substantially to the efficient development and delivery of safe, effective, and life-saving therapies to patients in need (EMA, 2018; ICH, 2020; FDA, 2019).
To sustain these crucial improvements and foster a culture of continuous improvement over the long term, continued and dedicated investment in cultivating a strong quality culture, providing comprehensive personnel development and training programs, and promoting seamless cross- departmental integration and collaboration will be absolutely necessary. These investments are not merely expenditures but rather strategic imperatives that underpin the long-term success and sustainability of biopharmaceutical organizations.
References
European Medicines Agency (EMA). (2025). Good Manufacturing Practice (GMP). Retrieved from https://www.ema.europa.eu/en/human-regulatory-overview/research-development/compliance- research-development/good-manufacturing-practice
International Council for Harmonisation (ICH). (2000). ICH Q7: Good Manufacturing Practice for Active Pharmaceutical Ingredients. Retrieved from https://www.ema.europa.eu/en/ich-q7-good- manufacturing-practice-active-pharmaceutical-ingredients-scientific-guideline
U.S. Food and Drug Administration (FDA). (2006). Guidance for Industry: Quality Systems Approach to Pharmaceutical Current Good Manufacturing Practice Regulations. Retrieved from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-systems- approach-pharmaceutical-current-good-manufacturing-practice-regulations






